Los bebés que se identifican en la prueba del talón (cribado metabólico) en el recién nacido, pueden comenzar el tratamiento antes de que comiencen los signos, y el 80-90% no desarrollarán ningún síntoma.
La gravedad de GA-1 varía mucho para cada individuo. Los síntomas y el tratamiento varían en diferentes personas con la misma enfermedad; generalmente comienzan en la infancia o la niñez temprana, pero a veces los síntomas comienzan en la adolescencia o la edad adulta y puede presentar muchos de estos signos o ninguno. En algunos casos, no desarrolla ningún síntoma, incluso si no recibe tratamiento, y solo se detectan después de diagnosticar a un hermano o hermana, o de forma casual.
Si GA1 no se trata, generalmente como resultado de una enfermedad aguda, una infección, fiebre, vacuna o intervención quirúrgica por lo general, el 80 ó 90% de los bebes desarrollarán trastornos neurológicos entre los 3 y 36 meses de edad; después de una descompensación metabólica causará una crisis encefalopática aguda, con daño grave de los ganglios basales, disminución del tono muscular (hipotonía), pérdida de las habilidades motoras y convulsiones que dan lugar a lesiones del núcleo estriado con distonía secundaria grave y en ocasiones hemorragia subdural y retiniana.
Los síntomas pueden ser difíciles de evaluar:
El estrés repetido en el cuerpo (como infección y fiebre) puede causar que los síntomas empeoren.
Algunos estudios han demostrado que la capacidad intelectual de una persona con GA1, incluso si no se trata, no se ve afectada.
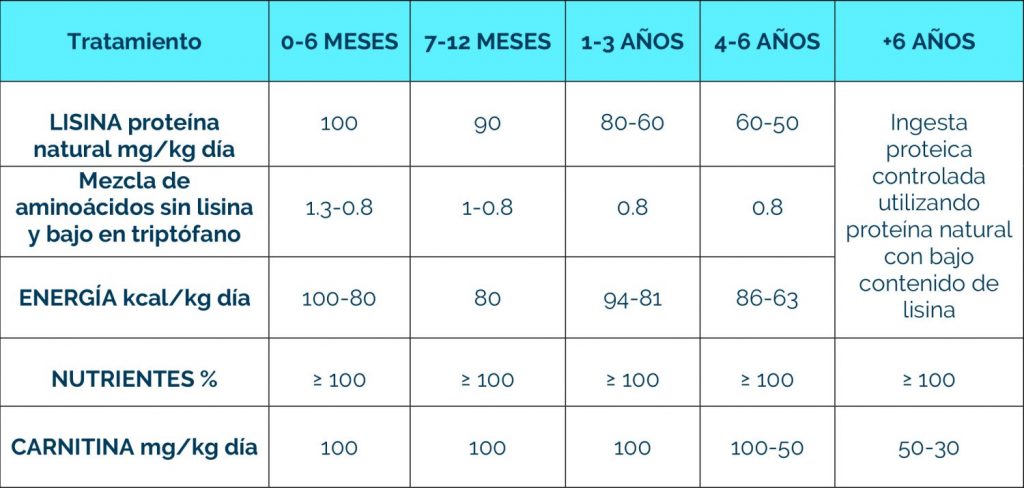
A los seis años de edad y con el tratamiento apropiado, el riesgo de crisis encefalopática disminuye.
En algunos pacientes, se desarrolla gradualmente una hipotonía y distonía sin ninguna crisis encefalopática, que es conocida como de aparición tardía o de aparición insidiosa. Pueden presentar síntomas neurológicos no específicos tales como cefaleas, vértigo, marcha atáxica transitoria, habilidades motoras finas reducidas, o desmayos después de hacer ejercicio, pero no desarrollan daño del núcleo estriado.
Recomendación número 3
Fuerte recomendación para: En niños con hemorragia Subdural (incluida la sospecha del síndrome de bebé sacudido) y / o colecciones de fluido bitemporal que sugieren hipoplasia frontotemporal y / o quistes aracnoideos, se recomienda un estudio diagnóstico utilizando el algoritmo para el estudio diagnóstico dirigido.
Nivel de evidencia Moderado SIGN 2+ a 4. Consistencia de evidencia moderada
Relevancia clínica Alta*1
Declaración 2:
El hallazgo de hemorragia subdural generalmente se acompaña de otras anormalidades radiológicas características de AG-I (por ejemplo hipoplasia frontotemporal, espacios de líquido cefalorraquídeo ensanchados etc). El hallazgo aislado de hemorragia subdural sin estas anormalidades características no es sugerente de AG-I per se y no debería llevar a un estudio diagnóstico dirigido. *1



")