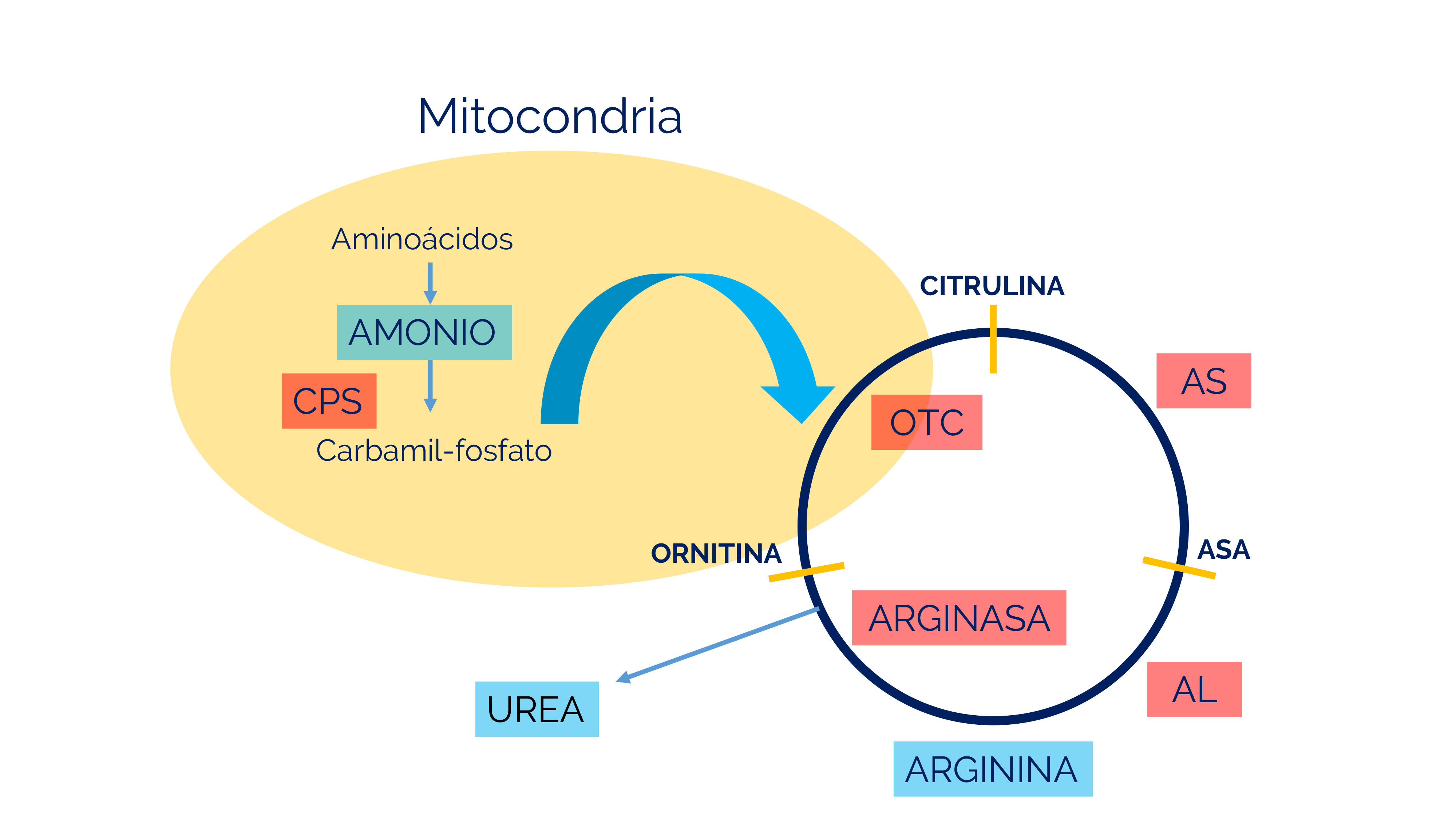
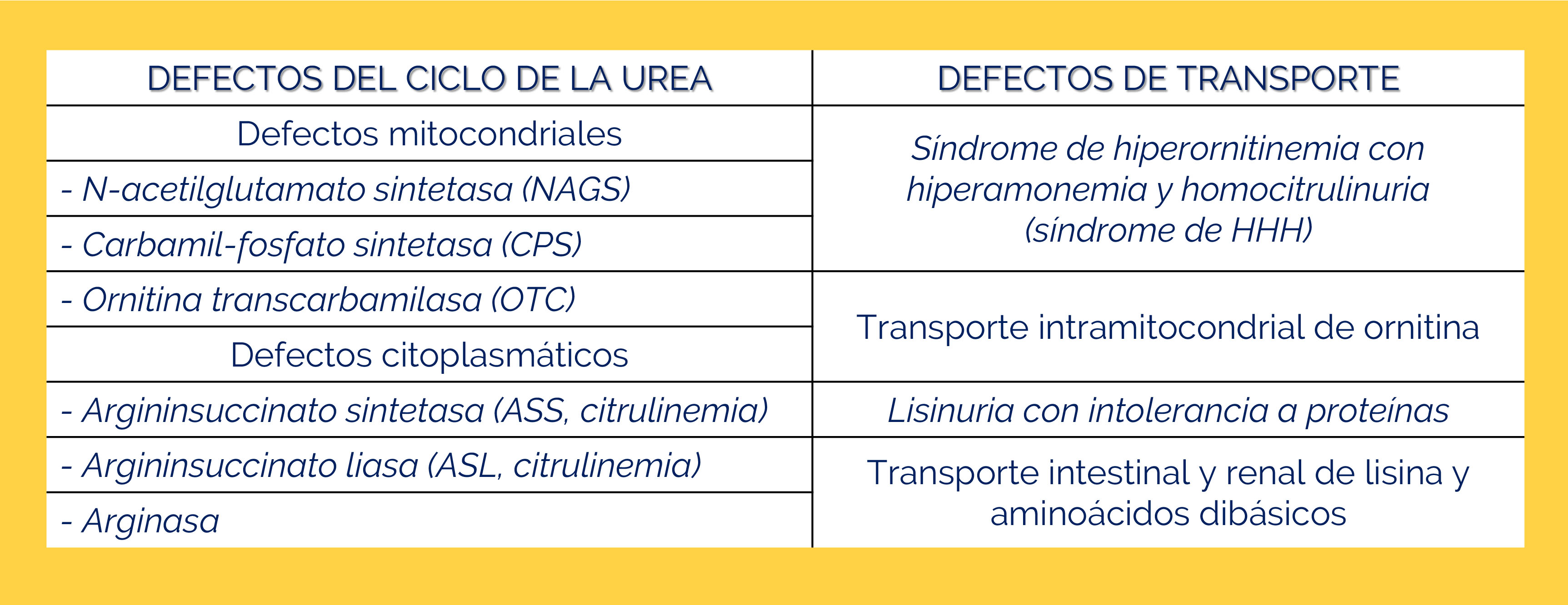
La enzima OTC combina el carbamilfosfato producido por CPS1 con un aminoácido llamado ornitina para producir citrulina.
Generalmente se sospecha en función de los síntomas y las pruebas bioquímicas, requiere pruebas genéticas o pruebas enzimáticas de una biopsia hepática para confirmar el diagnóstico.
La deficiencia de actividad de OTC causa hiperamonemia, generalmente es más bajo en las mujeres en comparación con los hombres.
Los pacientes con deficiencia OTC completa (el tipo más grave de este trastorno) desarrollan rápidamente altos niveles de amoníaco en la sangre, poco después del nacimiento y desarrollan síntomas en cualquier momento antes de una semana de edad. Los bebés que son tratados con éxito de esta primera crisis corren el riesgo de episodios repetidos de hiperamonemia.
El gen OTC se encuentra en el cromosoma X. Los hombres solo tienen un cromosoma X, mientras que las mujeres tienen dos. Por lo tanto, la mayoría de los pacientes con presentaciones graves de deficiencia de OTC son hombres. Las mujeres con un «gen OTC anormal» y un «gen OTC normal» pueden no mostrar ninguna evidencia clínica del trastorno; sin embargo, el 15% puede mostrar algunos síntomas o signos de la enfermedad. Los pacientes con deficiencia parcial de OTC (tipo de OTC más leve) pueden presentar en cualquier momento de la vida un evento desencadenante como una infección o estrés.
Si la madre es portadora de una mutación en el gen OTC, puede o no sufrir los efectos de la deficiencia enzimática.
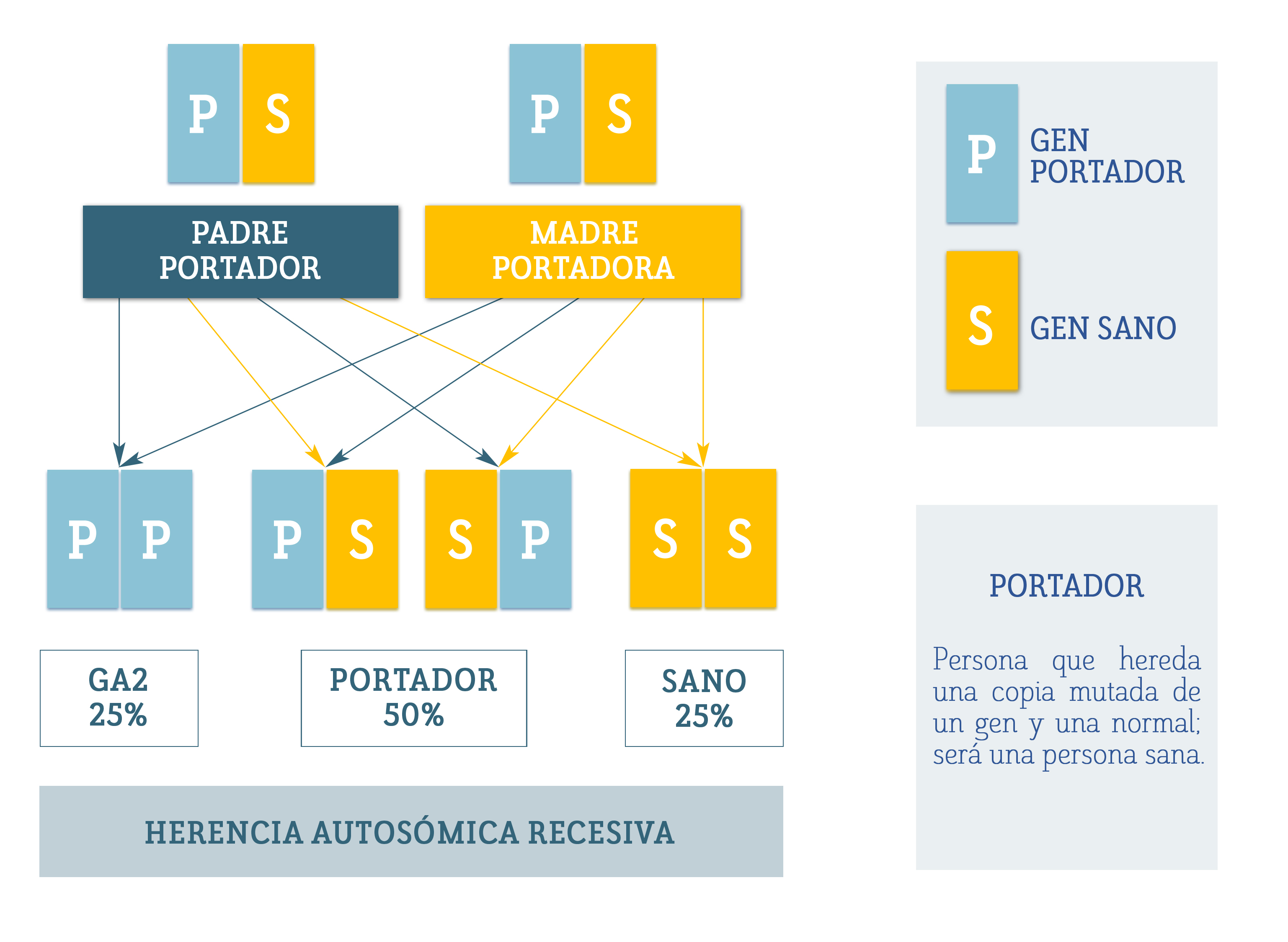
Si transmite la mutación a su hijo, pasándole su cromosoma X mutado, éste sufrirá la enfermedad, ya que solo posee un cromosoma X y éste contiene un gen OTC mutado.
Si lo transmite a una hija, que posee dos cromosomas X, ésta se convertirá en portadora, capaz de transmitir el gen mutado.
Es decir, el 50% de sus hijas serán portadoras y el 50% de sus hijos estarán afectados de deficiencia de OTC.
Existen también mutaciones espontáneas, no transmitidas por vía materna.
Los niños poseen un solo cromosoma X, heredado de su madre, ya que su padre le ha pasado un cromosoma Y, que determina su sexo masculino.
Las niñas poseen dos cromosomas X, uno heredado de su padre y uno de su madre. Si uno de ellos está mutado, la niña debería tener alrededor de un 50% de actividad enzimática residual de OTC.
No obstante, en las primeras etapas del desarrollo uno de los dos cromosomas X se inactiva al azar, pudiendo ser el que contiene el gen OTC mutado o el normal.
Todas las células que derivan de cada una de ellas tendrán las mismas características (mutadas o normales). Esto determina que el defecto de OTC se manifestará más o menos en hígado (órgano en el que se expresa la OTC) y la niña mostrará más o menos síntomas según el porcentaje de células hepáticas (hepatocitos) con el cromosoma X con la mutación activo.
Puede ser asintomática o mostrar una sintomatología variable, que se manifieste en la infancia, adolescencia o edad adulta o incluso una intoxicación aguda grave en el período neonatal si la expresión de la enzima OTC mutada fuera masiva en hígado.
En mujeres son más frecuentes las formas crónicas leves, con algún síntoma neurológico (retraso mental, ataxia, irritabilidad, agresividad, confusión, alucinaciones), digestivo (anorexia, intolerancia a proteínas) y hepático o bien las formas casi asintomáticas.
Si el padre tiene una mutación lo suficientemente leve para que le permita procrear, lo transmitirá solo a sus hijas (100% de hijas portadoras), junto con su cromosoma X, siendo todos sus hijos varones sanos.
El diagnóstico: clínica, amonio, aminoácidos y ácido orótico permiten el diagnóstico en los períodos de descompensación.
Las alteraciones bioquímicas pueden no ser obvias en mujeres portadoras en condiciones basales y habría que realizar un test de sobrecarga proteica o test de sobrecarga de alopurinol, para provocar una cierta descompensación controlada que permita el diagnóstico.
La confirmación se realiza mediante el estudio enzimático en biopsia hepática o duodenal (únicos tejidos en los que se expresa la OTC) y confirmación genética, que permiten el consejo genético y el diagnóstico prenatal.